Klebsiella pneumoniae Population Structure and Diversity
Dr. Kathryn Holt from the University of Melbourne published a very important paper in the Proceedings of the National Academy of Sciences yesterday, using whole genome sequencing to study the population structure as well as virulence and antimicrobial resistance factors of Klebsiella pneumoniae. A total of 288 isolates from 6 countries (Australia, Indonesia, Laos, Singapore, U.S.A. and Vietnam) were sequenced – 59 bovine isolates (26 carriage and 33 mastitis isolates), 9 environmental isolates, 4 monkey isolates, 2 sea mammal isolates, 2 plant isolates, 1 mouse isolate, with the rest being human isolates (community as well as hospital-acquired). As is typical of such papers, there is a lot of information that can only be found in the supplementary files, which is well worth the look. She has also put up the project in Microreact, from which I have obtained a screen capture. You can also read about her own thoughts of the study here.
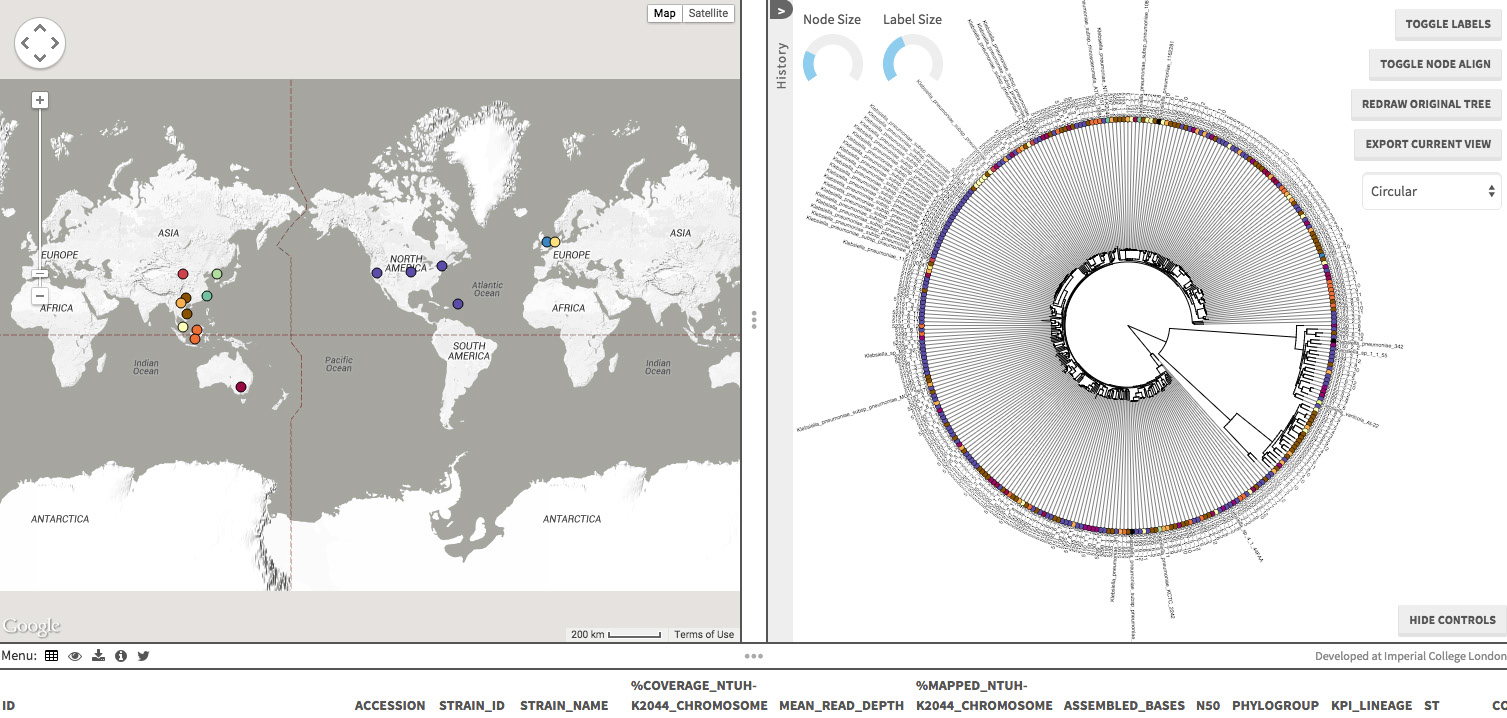
Screen capture from the Microreact site, detailing the Klebsiella pneumoniae project.
I am glad to be able to say that we in Singapore participated in the study, contributing 20 isolates including 6 from patients with liver abscess/endophthalmitis, a variety of nosocomial antimicrobial-resistant isolates, and even one obtained from sea water off East Coast Park in 2008. Our isolates were all from the Singapore General Hospital, kept by the Diagnostic Bacteriology chief blogger.
Most of us in the healthcare profession are only aware of K. pneumoniae as a human pathogen, associated with virulent infections in the community and antimicrobial-resistant infections in the hospital setting. However, their ecological distribution and impact is far greater. Using the data from whole genome sequencing, Dr. Holt’s team were able to support prior claims that the organism we know of as K pneumoniae should actually be 3 broad separate species (KPI, KPII and KPIII for convenience – KPI = Klebsiella pneumoniae, KPII = Klebsiella quasipneumoniae and KPIII = Klebsiella variicola).

The three distinct phylogroups of Klebsiella pneumoniae – screen capture of Figure 1A from the main PNAS manuscript.
KPI is the most broadly distributed, and is found in both plants, animals and humans. A significant minority harbour virulence genes (yersiniabactin and siderophores) and are associated with invasive community disease (i.e. liver abscess and endophthalmitis). KPII is found primarily in humans, and is associated with antimicrobial resistance. The vast majority do not possess siderophores and therefore are opportunistic pathogens, being associated with nosocomial disease. KPIII is found primarily in animals and plants, although some members can cause disease in both animals and humans.
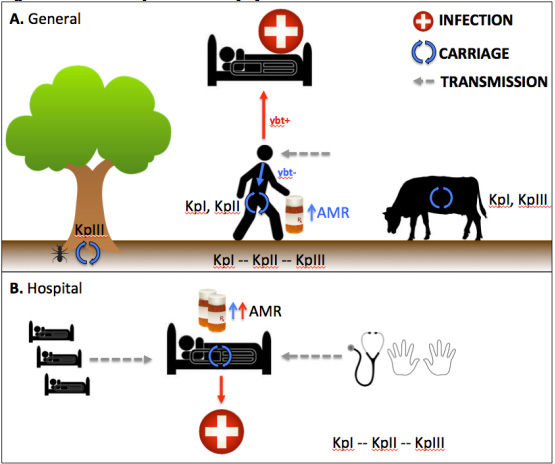
A supplemental figure (S12) from Dr. Kathry Holt’s manuscript in PNAS, showing the Klebsiella pneumoniae population.
Previous work had already linked several virulence factors and siderophores (small compounds secreted by bacteria – especially pathogenic bacteria – to bind iron, which are then taken back by the organism. Iron is essential for life in all organisms except viruses, and the ability to successfully obtain iron within the host organism despite the host’s efforts to “lock up” its iron stores, is crucial for survival and disease causation) to K. pneumoniae infection, and this is again strongly verified by the sequencing work. It is clear from the figure below that the presence of more siderophores is strongly associated with invasive infection (defined loosely as those infections where the organisms were isolated from a sterile site, as opposed to infections where organisms were obtained from non-sterile sites such as urine and sputum – hence perhaps the overlap).
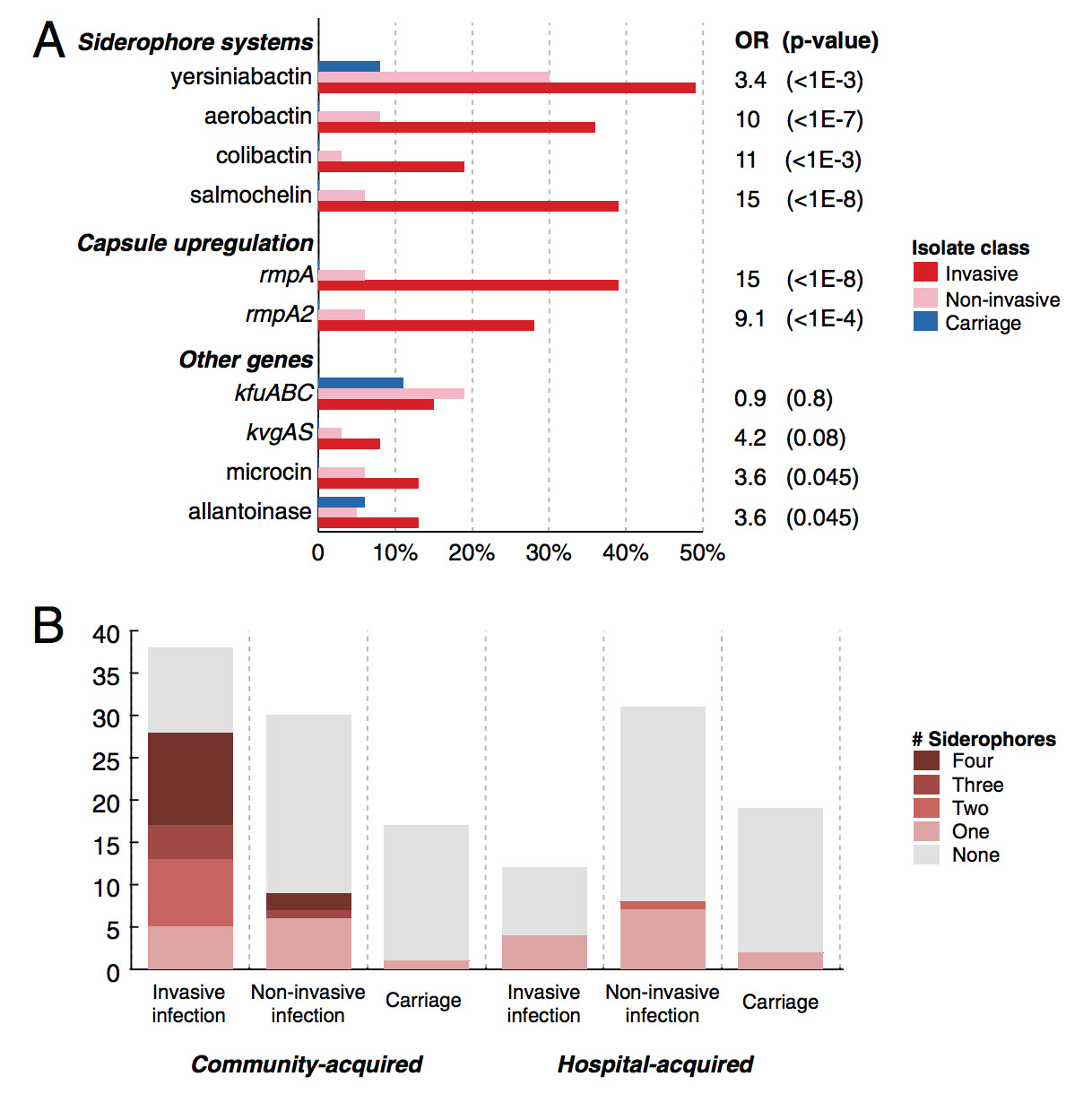
Virulence factors associated with Klebsiella pneumoniae infection. This is figure 3 from the main PNAS manuscript.
In cattle, however, the strains associated with bovine mastitis largely do not carry siderophores. Instead the majority of bovine mastitis strains have an acquired lac operon gene cluster that permits the organism to utilise lactose (from milk). Makes sense, naturally!
I had previously stated that unlike “classical” strains, “hypervirulent” strains of K. pneumoniae are rarely associated with antimicrobial resistance, and this is also largely true here. However, there are isolates within the large collection that suggest – rather worryingly – that there is a possible convergence of virulence genes with antimicrobial resistance. This seems to be the case for the epidemic clonal complex 258 (CC258) K. pneumoniae that is also associated with one of the carbapenemase (KPC) genes – several of these isolates were also carrying the yersiniabactin virulence gene.
What about the isolates from Singapore in this study? They were virtually all KPI’s except for a KPII from sea water and another KPII causing a nosocomial catheter-related urinary tract infection. The most common nosocomial sequence type (ST) was ST11, which globally is associated with carriage of extended-spectrum beta-lactamase (ESBL) genes. All six community isolates causing invasive infection were from ST’s that had been associated with invasive disease in the region, especially ST23 (3 isolates – 2 from patients with liver abscess and one from a patient with necrotising fasciitis of the thigh). Singapore as a “global city” is a perspective that can also be appreciated via analysis of its various disease-causing pathogens.
There is an intriguing possibility raised by work such as this – as microbial genomic analysis becomes more routine (as well as faster and cheaper!), it will have the potential to transform clinical practice substantially. Besides antimicrobial susceptibility data, for which conventional microbiology laboratory methods work well enough, the presence or absence of virulence genes in K. pneumoniae (and possibly other pathogens) may well dictate how extensive the work-up for the patient should be (i.e. should one get a CT scan to look for liver abscess, or eye screening to look for endophthalmitis, etc.) as well as potentially antibiotic selection and treatment duration.
